North References for Navigating with Map, Compass and GPS
To use a compass with your map and your GPS you need to have an understanding of the north reference used by each of them. There are three different norths in common use and in some cases choices to be made about which to use on your compass, map and GPS.
The three options for north
True North

The True North Pole is the axis of the earth’s rotation. The North Star is used as a true north reference. It’s position in the sky causes it to appear almost stationary with the other stars rotating around it. Lines or meridians of longitude can also be used as true north reference lines. Meridians of longitude converge at the True North and South Poles. The vertical edges of many maps are defined by a meridian of longitude, and can be used as a true north reference.
Magnetic North

The magnetic poles are aligned with the earth’s magnetic field. A free floating magnetic needle in a compass will align itself with the magnetic field and thus points to the magnetic poles. Declination is the angular difference between true north and magnetic north for a given location. The Magnetic Pole may appear to be either east or west of the True North Pole. Declination changes depending on your position relative to the two poles. Declination also changes over time, because the location of the magnetic poles changes with time. Most GPS receivers will calculate the declination for their current position. This is often referred to as “automatic” north reference.
Grid North

Grid north is useful because it allows you to use the UTM grid lines on your map as your north reference. Grid north is typically within 2° east or west of true north and varies with your position within a UTM zone. When minimal accuracy is all that is required, it is common to treat grid north lines as true north lines without accounting for the small difference.
Declination Diagram
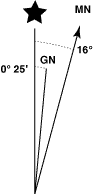
Most paper maps use a printed declination diagram to show the relative direction of the three north references. Typically, there will be an arrow that point to the top of the map and is parallel to the edges of the map. This is the "top of the map" north reference that was used when the map was printed. Commonly it will be either True North (sometimes designated with a star, ot the letters TN) or Grid North (often designated with the letters GN). The angles between the three north references will be printed on the diagram.
The diagram should also have a date that the angle between True North and Magnetic North was determined. This declination angle changes over time. You should use a current value, not a many years old value printed on an old map. A source for the current declination at a given location is the National Geophysical Data Center's declination calculator. Your GPS can also calculate the declination for your current location.
Some maps have only a North Arrow, which may or may not indicate which north it is referencing. In some cases north on the map may not be at the top of the sheet. To use these maps with your compass and GPS, you will need to seek additional clues as to the north reference. You will also need to use a declination calculator to determine the current declination angle.
Customized North Reference Sheet
I've recently added a new page that allows you to look up the current declination for your location, and input the north reference choices you have made for your map and compass. The page will create a customized reference card for your specific situation. Change the settings and the diagram will change as well. It's a great way to play with various north reference choices and to learn who they impact the work you do in the field.
Design your own declination reference sheet.

Choosing a North Reference, for your Map, your Compass, and your GPS
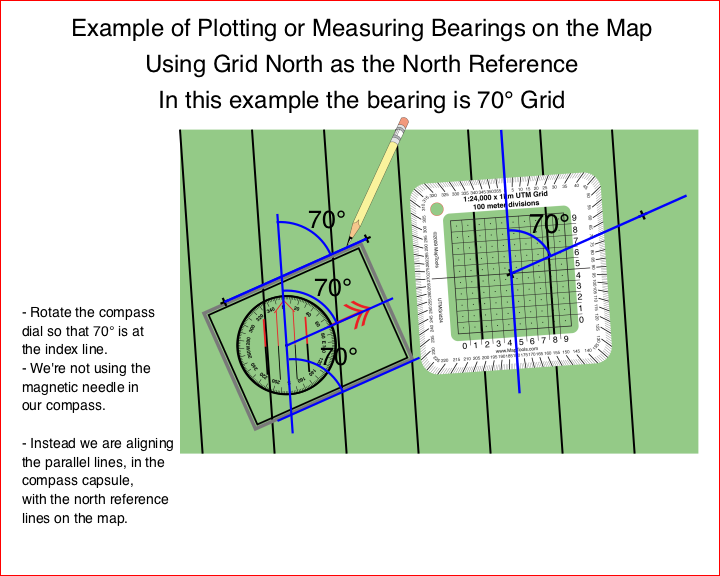
You may have choices to make between north references on your map and your compass. Your GPS can be set to use any of the three common references. When the north references are different between your map, compass, and GPS, you will need to do conversions as you move bearings between them.
North Reference on your Map
Grid North
Grid North is easy to use on maps with printed UTM / MGRS / USNG grid lines.
- Lots of north reference lines already printed on the map.
- Likely to be very close to True North.
- When the level of accuracy required is low, Grid North lines are often used as True North lines.
True North
Lines of Longitude are True North lines. Often the two vertical edges of the map are lines of longitude and can be used as True North reference lines. They may be the only two True North lines on you map, unless your map has a lat/lon grid printed on it.
Magnetic North
Many aviation and marine charts have pre-printed Magnetic North lines. Most other maps do not. You can draw parallel lines aligned with Magnetic North onto your map for use as north reference lines.
North Reference on your Compass
Magnetic North
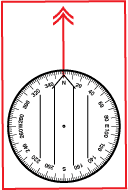
The needle or card of your compass will always align itself with Magnetic North. Thus, Magnetic North is an easy and natural choice to use with your compass
A compass that has not been adjusted to a different north reference, will read zero degrees when it is aligned with Magnetic North. All bearings taken with this compass should be referred to as “magnetic bearings.” Since the compass is a magnetic device this is an intuitive result. It is also the result provided by compasses that cannot be adjusted. To plot a magnetic bearing onto your map you will likely need to convert the bearing to either a True or Grid North reference by adding or subtracting the declination for your current location. There is no need to change the adjustment based on your location.
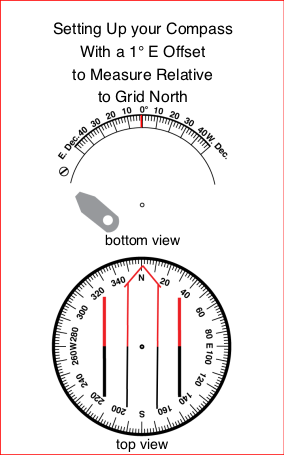
On a compass that reads relative to Magnetic north, the orientation arrow on the baseplate will align with the 0° or North mark on the dial, as is shown to the right.
Adjusting the North Reference on Your Compass to True or Grid North
Many compasses allow you to adjust the position of the lines used to align the magnetic needle with respect to the angle measuring dial. The adjustment mechanism is typically either a gear driven one with a small slotted screw and a small brass screwdriver on the lanyard, or a simple friction fit between the capsule and the dial. The choice to adjust the north reference on your compass will affect how you work with your compass and should be considered carefully.
Regardless of your decision on whether to adjust the north reference or not, you must occasionally check to see that the compass is set as you expect it to be. This is particularly true for the friction fit mechanisms which may loosen with time and adjust themselves.
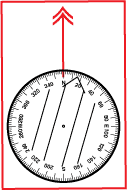
A compass that is adjusted will resemble the declination diagram. The orienting arrow and the parallel lines in the bottom of the capsule will be aligned with Magnetic North, not the 0° or North mark on the dial. The zero degree mark on the dial is aligned with either True or Grid North. All bearings taken with this compass should be referred to as either “true bearings” or “grid bearings” depending on the north reference you have adjusted to. You will likely not need to add or subtract conversion values to plot the bearing onto your map. You will need to remember to change the adjustment to match your current location.
When the orientation arrow can not be adjusted independently of the angular dial, you can use some other mark to align the compass needle as pictured below. Some compasses have a printed scale for this purpose.



Card style compasses and sighting compasses generally can not be adjusted, and will always provide bearings relative to Magnetic North. This is because the card and magnet are fixed to each other and sealed inside the capsule.
A common problem for beginners occurs when someone adjusts their compass for them, and then they forget what was done and why. An incorrectly adjusted compass is useless for anything beyond a general sense of direction and may be worse than no compass at all.
Two Schools of Thought
Set your compass and forget it Adjust for the declination in your compass.
- All bearings will be Grid or True.
- No conversion required to use it on a map.
- Don’t forget to check the setting occasionally.
- Don’t forget to change it when you go somewhere else.
Set your compass to 0°, and always think about it
- All bearings will be Magnetic.
- Conversion to Grid or True, or drawing Magnetic North reference lines on your map, will be necessary for map work.
- Works with all compasses.
- You are more likely to remember how declination works.
- If your compass is adjustable, don’t forget to check the setting occasionally.
North Reference on your GPS
You GPS can be set to use any of the three north references. The north reference choice is usually located in the setup page for Heading or North Reference. Some GPS receivers use the name "Automatic" to indicate that the GPS should calculate the declination for the current location and display directional information relative to Magnetic North. There is often a choice that allows the user to enter a declination value to be used. This is probably not what you want. Make sure you understand why you are using a user defined declination if you choose to do so.
Converting Between North References
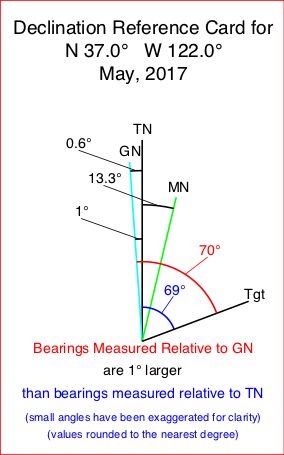
The key to converting between the three north references is to add a line representing the bearing to an imaginary target to the declination diagram. Now it is easy to see the different angles measured to the target from each of the north references. Remember the angle is measured from the desired north reference line to the target bearing line. The measured angle’s zero degree value is associated with the north reference line and increases in a clockwise direction.
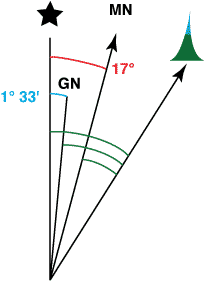
In the example at the right, both Magnetic and Grid North are east of True North. A true bearing would be 17° larger than a magnetic bearing. Thus, to convert from a magnetic bearing to a true bearing you would add 17°.
The angle measured from the target to Grid North is also larger than the angle measured from the target to Magnetic North. The difference is the 17° angle from True North to Magnetic North less the 1° 33’ angle from True North to Grid North. Thus, to convert from a magnetic bearing to a Grid North reference you would add 15° 27’. (I would likely round this to 15 1/2°. I might even round to 15° if accuracy was not critical.)
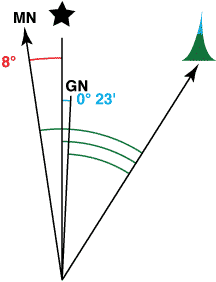
In the example at the right, both Magnetic North is west of True North and Grid North is east of True North. A true bearing would be 8° smaller than a magnetic bearing. Thus, to convert from a magnetic bearing to a true bearing you would subtract 8°.
The angle measured from the target to Grid North is also smaller than the angle measured from the target to Magnetic North. The difference is the 8° angle from True North to Magnetic North plus the 0° 23’ angle from True North to Grid North. Thus, to convert from a magnetic bearing to a Grid North reference you would subtract 8° 23’. (I would likely round this to 8 1/2°or just 8°.)
Common Scenarios
Map=Grid, Compass=Mag, GPS=Mag
- Easy to use reference lines already on the map.
- No compass adjustment needed.
- Conversion between Grid and Magnetic is required to work with compass bearings on the map.
Map=Grid, Compass=Grid, GPS=Grid
- Easy to use reference lines already on the map.
- Compass adjusted to Grid North.
- Adjustment should be checked for correctness
- No conversions required to work with compass bearings on the map.
Map=Mag, Compass=Mag, GPS=Mag
- You will need to draw reference lines on your maps.
- No compass adjustment needed.
- No conversions required to work with compass bearings on the map.
- You only need to worry about north reference at home when you draw the lines on your maps.
An All too Common Scenario
All too often I when encounter someone who is having trouble working with their map and compass they tell me, "Someone else set my compass up for me." I ask some question...
- Is it set for true, grid, or magnetic bearings? I don’t remember.
- Is it set for the declination here, or somewhere else? I don’t know.
- Have you checked that it is set correctly? I don’t know how to check it, or how to adjust it.
What Do I Use
My maps are gridded for UTM coordinates. So I use Grid North on the map. I use a sighting compass that con not be adjusted. So I use Magnetic North on my compass. When I move a bearing between my compass and my map, I convert the north reference. My GPS is set to automatically calculate the declination and give directions relative to Magnetic North. When I goto a waypoint on the GPS, the bearing to the waypoint is magnetic. I can grab my compass, turn to face the bearing and be facing the target. Yes my GPS has a compass in it, and I could use it to face the target as well. But without any sighting mechanism, just using the arrow on the screen is not particularly accurate.
No bearing or heading is complete without the word
True, Magnetic, or Grid following it.
Don’t make people guess, say it and write it!
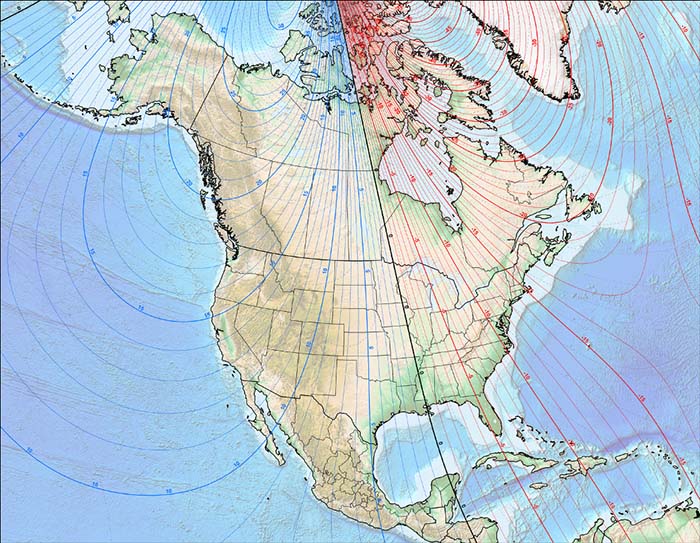
2010 North American Magnetic Declination
Current declination maps and calculators can be found at the National Geophysical Data Center's website.
I have a 4-page color handout on North references (single sheet folded in half) that I use in my navigation classes. I will send you one for free with you order if you wish. Or you can download the pdf file and take a look. If you want to use the handout for your class, you can order them for less than it would cost you to print them on your own printer.
| Item description | Price |
|
North Reference Sheet Part Id: NorthRefSheet, Made in US, UPC: 824249001211 |
$0.00 |